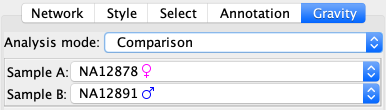
Comparison of 2 samples¶
In this mode you will analyse two samples. It is mostly targetting people interested in somatic variants and could allow to compare germline versus somatic, or different time point, or different tissues.
In the control panel you must first select the two sample to analyse.
Network tab¶
The network tab (1 in figure below) of the control panel contains 2 parts:
- the Interactions used to create the network
- the pathway of gene lists that will be displayed in the network
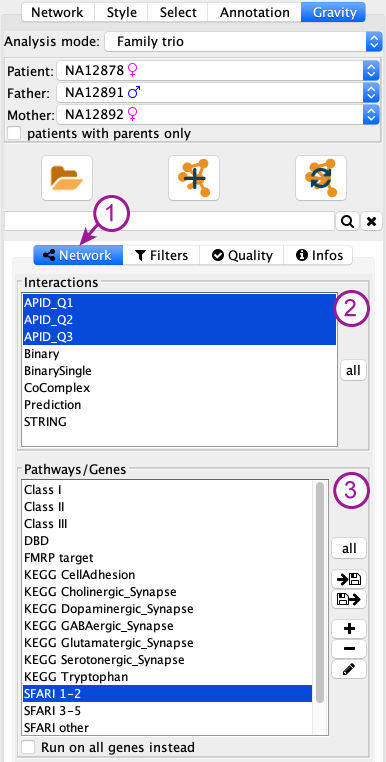
Interactions (2)¶
By default we are providing different protein-protein interactions networks.
- APID [ALONSO-LOPEZ2016]
- STRING [SZKLARCZYK2015]
- PPI maps from Rolland et al. [ROLLAND2014]
- a systematic co-complex PPI map [HUTTLIN2015]
Pathway / genes (3)¶
By default we are providing several gene lists and pathways of interest for brain associated diseases such as:
- Synaptic pathways from the KEGG database [KANEHISA2016]
- Class I-III genes [YUEN2015]
- TADA genes [SANDERS2015]
- SFARI genes [ABRAHAMS2013]
- FMRP targets [DARNELL2011]
- Developmental Brain Disorder genes [GONZALEZ-MANTILLA2016]
The user can easily create, edit, or remove list of genes. A gene list consists of a title and a list of Gene SYMBOL (one per line).
Filters¶
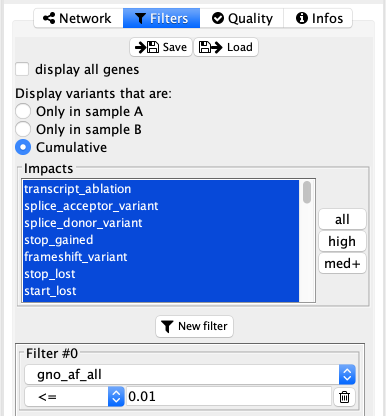
The very first option in this tab is a simple checkbox display all genes. When unselected, the behaviour will be to display on genes carrying variants. When selected, the network will display all the genes from the selected gene lists and pathway from the network tab. This option can be very important in several cases:
- if you want to see interactions inside a pathway, if you do not check it, you will only see direct interactions between mutated genes. (no intereaction of mutated gene through a non-mutated gene)
- if you want to perform a burden analysis on a pathway or a set of gene, you must check this box.
This tab allow to filter the data on multiple criteria such as:
Mode of comparison¶
In the comparison mode, this section is different and allow to select what the user wants to observed. The three options are variants that are found only in sample A, B or either of the samples (cumulative). The cumulative mode could help finding interactions between somatic and germline variants.
Impacts¶
Here it is possible to quickly filter based on the functional impact annotated by VEP. 3 quick select buttons ae available on the right side of the list:
- all: select all impacts in the list
- high: select impacts with a severity of HIGH for VEP, which corresponds to loss-of-function variants
- med+: select impacts with a severity of HIGH or MEDIUM for VEP, which adds to the previous category variants such as missense, inframe insertion/deletions, or splice region variants
Other filters¶
The last part allows to filter on attributes from the VCF INFO fields (or GEMINI columns). Attributes types are interpreted using the VCF header. All filters condition must be fullfilled (logical AND between filters).
Quality¶
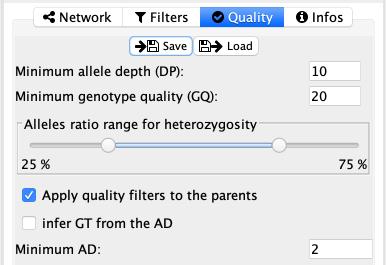
The quality tab allows for quality filtering on the variants, based on the depth (DP), the genotyping quality (GQ) and the allelic ratio (alternate allele depth over total depth). This way it is possibly to restrict display variants to higher quality ones. The allelic ratio particularly avoids sequencing errors in lower coverage sections and mosaicism.
It is possible for the user to check if the parents must fulfill the same quality filters or not. This could be important to obtain high quality variants for de novo mutations. But it must be noted that variant that are not passing the filters are not displayed at all.
The last two options are more specific for people working on somatic mutations that were already cleaned in their pipeline. By checking the infer GT from the AD and setting the Minimum AD value, the user can set the AD threshold after which the variant is considered present.
References¶
| [ALONSO-LOPEZ2016] | Alonso-López, D., et al. “APID interactomes: providing proteome-based interactomes with controlled quality for multiple species and derived networks.” Nucleic Acids Research. 44 (2016): W529-W535. |
| [SZKLARCZYK2015] | Szklarczyk, D., et al. “STRING v10: protein-protein interaction networks, integrated over the tree of life.” Nucleic Acids Research. 43 (2015): D447-D452. |
| [ROLLAND2014] | Rolland, T., et al. “A Proteome-Scale Map of the Human Interactome Network.” Cell. 159 (2014): 1212-1226. |
| [HUTTLIN2015] | Huttlin, E.L., et al. “The BioPlex Network: A Systematic Exploration of the Human Interactome.” Cell. 162 (2015): 425-440. |
| [KANEHISA2016] | Kanehisa, M., et al. “KEGG as a reference resource for gene and protein annotation.” Nucleic Acids Research. 44 (2016): D457-D462. |
| [YUEN2015] | Yuen, R.K.C., et al. “Whole-genome sequencing of quartet families with autism spectrum disorder.” Nature Medicine. 21 (2015): 185. |
| [SANDERS2015] | Sanders, S.J., et al. “Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci.” Neuron. 87 (2015): 1215-1233. |
| [ABRAHAMS2013] | Abrahams, B.S., et al. “SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs).” Molecular Autism. 4 (2013): 36. |
| [DARNELL2011] | Darnell, J.C., et al. “FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism.” Cell. 146 (2011): 247-261. |
| [GONZALEZ-MANTILLA2016] | Gonzalez-Mantilla, A.J., et al. “A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders.” JAMA Psychiatry. 73 (2016): 275-283. |