Controls¶
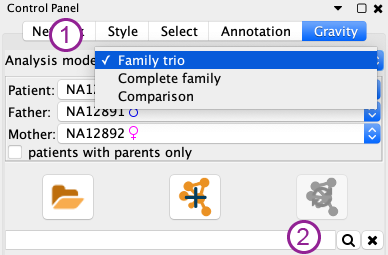
In the control panel, it is possible to select the analysis mode (1 in the Figure above).
- Family trio: made to analyse an individuals with its parents (missing parents are possible, it will just give less information)
- Complete family: made to analyse an entire familial pedigree to study segregation in a family.
- Comparison of 2 samples: to visualise what is shared or specific to each of the 2 samples (e.g. germline/somatic, before/after treatment).
Those 3 modes are describe in the following parts of this documentation.
In this part of the control panel, shared by all analysis mode, is a quicksearch field (2 in Figure above). It is possible to enter gene symbols (comma, semicolon or space separated) and also to use wildcards (asterisk) at the end of gene names. For instance, CNTN* will return a network of all the contactin genes. Running a search will create a new network restricted to the searched genes, all the currently selected filters are applied, and it will necessarily display all the searched genes, even if they do not carry any variant.
Note
The values in the search field are not used by the create a new network and update the current network buttons, but only by the small search button next to the search field itself or by pressing enter from it.